Correlation to nuclear segmentation#
The nuclear_correlation module provides metrics to assess how well cell segmentation masks align with nuclear segmentation masks. It is applicable only when nuclear masks are available.
[1]:
%load_ext autoreload
%autoreload 2
[2]:
import spatialdata as sd
sdata = sd.read_zarr("/home/lazic/src/segtraq/tests/data/xenium_sp_subset.zarr")
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/dask/dataframe/__init__.py:31: FutureWarning: The legacy Dask DataFrame implementation is deprecated and will be removed in a future version. Set the configuration option `dataframe.query-planning` to `True` or None to enable the new Dask Dataframe implementation and silence this warning.
warnings.warn(
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/xarray_schema/__init__.py:1: UserWarning: pkg_resources is deprecated as an API. See https://setuptools.pypa.io/en/latest/pkg_resources.html. The pkg_resources package is slated for removal as early as 2025-11-30. Refrain from using this package or pin to Setuptools<81.
from pkg_resources import DistributionNotFound, get_distribution
version mismatch: detected: RasterFormatV02, requested: FormatV04
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/zarr/creation.py:614: UserWarning: ignoring keyword argument 'read_only'
compressor, fill_value = _kwargs_compat(compressor, fill_value, kwargs)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/zarr/creation.py:614: UserWarning: ignoring keyword argument 'read_only'
compressor, fill_value = _kwargs_compat(compressor, fill_value, kwargs)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/zarr/creation.py:614: UserWarning: ignoring keyword argument 'read_only'
compressor, fill_value = _kwargs_compat(compressor, fill_value, kwargs)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/zarr/creation.py:614: UserWarning: ignoring keyword argument 'read_only'
compressor, fill_value = _kwargs_compat(compressor, fill_value, kwargs)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/zarr/creation.py:614: UserWarning: ignoring keyword argument 'read_only'
compressor, fill_value = _kwargs_compat(compressor, fill_value, kwargs)
version mismatch: detected: RasterFormatV02, requested: FormatV04
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/zarr/creation.py:614: UserWarning: ignoring keyword argument 'read_only'
compressor, fill_value = _kwargs_compat(compressor, fill_value, kwargs)
version mismatch: detected: RasterFormatV02, requested: FormatV04
version mismatch: detected: RasterFormatV02, requested: FormatV04
[3]:
sdata
[3]:
SpatialData object, with associated Zarr store: /g/huber/users/lazic/src/segtraq/tests/data/xenium_sp_subset.zarr
├── Images
│ ├── 'he_image': DataTree[cyx] (3, 448, 527), (3, 224, 264), (3, 112, 131), (3, 56, 66), (3, 28, 33)
│ ├── 'if_image': DataTree[cyx] (3, 221, 260), (3, 111, 130), (3, 55, 65), (3, 28, 33), (3, 13, 16)
│ ├── 'morphology_focus': DataTree[cyx] (1, 763, 899), (1, 381, 450), (1, 191, 225), (1, 95, 112), (1, 47, 56)
│ └── 'morphology_mip': DataTree[cyx] (1, 763, 899), (1, 381, 450), (1, 191, 225), (1, 95, 112), (1, 47, 56)
├── Labels
│ ├── 'cell_labels': DataTree[yx] (763, 899), (381, 450), (191, 225), (95, 112), (47, 56)
│ └── 'nucleus_labels': DataTree[yx] (763, 899), (381, 450), (191, 225), (95, 112), (47, 56)
├── Points
│ └── 'transcripts': DataFrame with shape: (<Delayed>, 8) (3D points)
├── Shapes
│ ├── 'cell_boundaries': GeoDataFrame shape: (158, 1) (2D shapes)
│ ├── 'cell_circles': GeoDataFrame shape: (122, 2) (2D shapes)
│ └── 'nucleus_boundaries': GeoDataFrame shape: (132, 1) (2D shapes)
└── Tables
└── 'table': AnnData (122, 313)
with coordinate systems:
▸ 'global', with elements:
he_image (Images), if_image (Images), morphology_focus (Images), morphology_mip (Images), cell_labels (Labels), nucleus_labels (Labels), transcripts (Points), cell_boundaries (Shapes), cell_circles (Shapes), nucleus_boundaries (Shapes)
[4]:
sdata["table"].obs
[4]:
| cell_id | transcript_counts | control_probe_counts | control_codeword_counts | total_counts | cell_area | nucleus_area | region | |
|---|---|---|---|---|---|---|---|---|
| 4612 | 4613 | 210 | 0 | 0 | 210 | 272.066406 | 27.319531 | cell_boundaries |
| 4613 | 4614 | 192 | 0 | 0 | 192 | 154.208594 | 33.280156 | cell_boundaries |
| 4614 | 4615 | 174 | 0 | 0 | 174 | 170.193906 | 31.744844 | cell_boundaries |
| 4615 | 4616 | 70 | 0 | 0 | 70 | 85.796875 | 26.235781 | cell_boundaries |
| 4616 | 4617 | 85 | 0 | 0 | 85 | 101.737031 | 16.843281 | cell_boundaries |
| ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 80758 | 80759 | 227 | 0 | 0 | 227 | 256.984219 | 42.221094 | cell_boundaries |
| 80759 | 80760 | 198 | 0 | 0 | 198 | 222.800938 | 30.028906 | cell_boundaries |
| 80760 | 80761 | 110 | 0 | 0 | 110 | 111.355313 | 33.144688 | cell_boundaries |
| 80761 | 80762 | 150 | 0 | 0 | 150 | 322.189844 | 60.644844 | cell_boundaries |
| 80762 | 80763 | 109 | 0 | 0 | 109 | 152.763594 | 41.769531 | cell_boundaries |
122 rows × 8 columns
The spatialdatadataset contains cell and nuclear masks as Shapesand Labels, as shown below.
Intersection over Union between cell and nucleus masks#
First, we compute the Intersection over Union (IoU) between cell and nuclear masks using compute_cell_nuc_ious.
[5]:
import time
import segtraq as st
n = 1
start = time.time()
results_df = st.nc.compute_cell_nuc_ious(sdata, n_jobs=n)
end = time.time()
print(f"Elapsed time with {n} threads: {end - start:.2f} seconds")
Processing IoU between cells and nuclei: 100%|██████████| 158/158 [00:00<00:00, 702.68it/s]
Elapsed time with 1 threads: 0.23 seconds
For each cell_id, we obtain the ID (best_nuc_id) of the nucleus mask with the highest IoU. If a cell does not overlap with any nucleus, the function returns a missing value for best_nuc_id. If the nucleus has an invalid geometry, IoUis reported as NA.
[6]:
results_df
[6]:
| cell_id | best_nuc_id | IoU | |
|---|---|---|---|
| 0 | 4613 | 4613 | 0.094093 |
| 1 | 4614 | 4614 | 0.207238 |
| 2 | 4615 | 4615 | 0.178686 |
| 3 | 4616 | 4616 | 0.292909 |
| 4 | 4617 | 4617 | 0.161824 |
| ... | ... | ... | ... |
| 153 | 80765 | None | 0.000000 |
| 154 | 80766 | None | 0.000000 |
| 155 | 80767 | None | 0.000000 |
| 156 | 80768 | None | 0.000000 |
| 157 | 80769 | None | 0.000000 |
158 rows × 3 columns
We will store the results in the .obs of the Table within the sdata object for plotting.
[7]:
sdata["table"]
[7]:
AnnData object with n_obs × n_vars = 122 × 313
obs: 'cell_id', 'transcript_counts', 'control_probe_counts', 'control_codeword_counts', 'total_counts', 'cell_area', 'nucleus_area', 'region'
var: 'gene_ids', 'feature_types', 'genome'
uns: 'spatialdata_attrs'
obsm: 'spatial'
[8]:
ious_df = results_df.set_index("cell_id")
sdata["table"].obs["IoU"] = sdata["table"].obs["cell_id"].map(ious_df["IoU"])
sdata["table"].obs["best_nuc_id"] = sdata["table"].obs["cell_id"].map(ious_df["best_nuc_id"])
[9]:
sdata
[9]:
SpatialData object, with associated Zarr store: /g/huber/users/lazic/src/segtraq/tests/data/xenium_sp_subset.zarr
├── Images
│ ├── 'he_image': DataTree[cyx] (3, 448, 527), (3, 224, 264), (3, 112, 131), (3, 56, 66), (3, 28, 33)
│ ├── 'if_image': DataTree[cyx] (3, 221, 260), (3, 111, 130), (3, 55, 65), (3, 28, 33), (3, 13, 16)
│ ├── 'morphology_focus': DataTree[cyx] (1, 763, 899), (1, 381, 450), (1, 191, 225), (1, 95, 112), (1, 47, 56)
│ └── 'morphology_mip': DataTree[cyx] (1, 763, 899), (1, 381, 450), (1, 191, 225), (1, 95, 112), (1, 47, 56)
├── Labels
│ ├── 'cell_labels': DataTree[yx] (763, 899), (381, 450), (191, 225), (95, 112), (47, 56)
│ └── 'nucleus_labels': DataTree[yx] (763, 899), (381, 450), (191, 225), (95, 112), (47, 56)
├── Points
│ └── 'transcripts': DataFrame with shape: (<Delayed>, 8) (3D points)
├── Shapes
│ ├── 'cell_boundaries': GeoDataFrame shape: (158, 1) (2D shapes)
│ ├── 'cell_circles': GeoDataFrame shape: (122, 2) (2D shapes)
│ └── 'nucleus_boundaries': GeoDataFrame shape: (132, 1) (2D shapes)
└── Tables
└── 'table': AnnData (122, 313)
with coordinate systems:
▸ 'global', with elements:
he_image (Images), if_image (Images), morphology_focus (Images), morphology_mip (Images), cell_labels (Labels), nucleus_labels (Labels), transcripts (Points), cell_boundaries (Shapes), cell_circles (Shapes), nucleus_boundaries (Shapes)
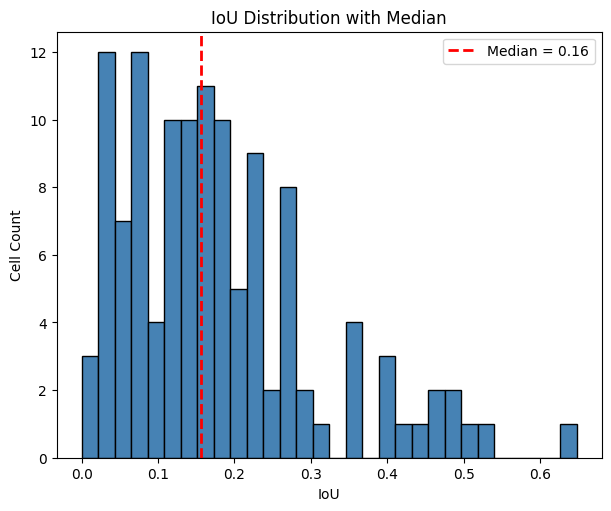
The histogram of IoU distribution (below) shows a median IoU is at 0.16, i.e. 16% overlap.
[10]:
import matplotlib.pyplot as plt
import numpy as np
# Prepare IoU values
ious = sdata["table"].obs["IoU"].dropna()
median_iou = np.median(ious)
# Set up figure with histogram
fig, ax = plt.subplots(figsize=(6, 5), constrained_layout=True)
# Plot histogram
ax.hist(ious, bins=30, color="steelblue", edgecolor="black")
# Add median line
ax.axvline(
median_iou,
color="red",
linestyle="--",
linewidth=2,
label=f"Median = {median_iou:.2f}",
)
# Decorate
ax.set_title("IoU Distribution with Median")
ax.set_xlabel("IoU")
ax.set_ylabel("Cell Count")
ax.legend()
plt.show()
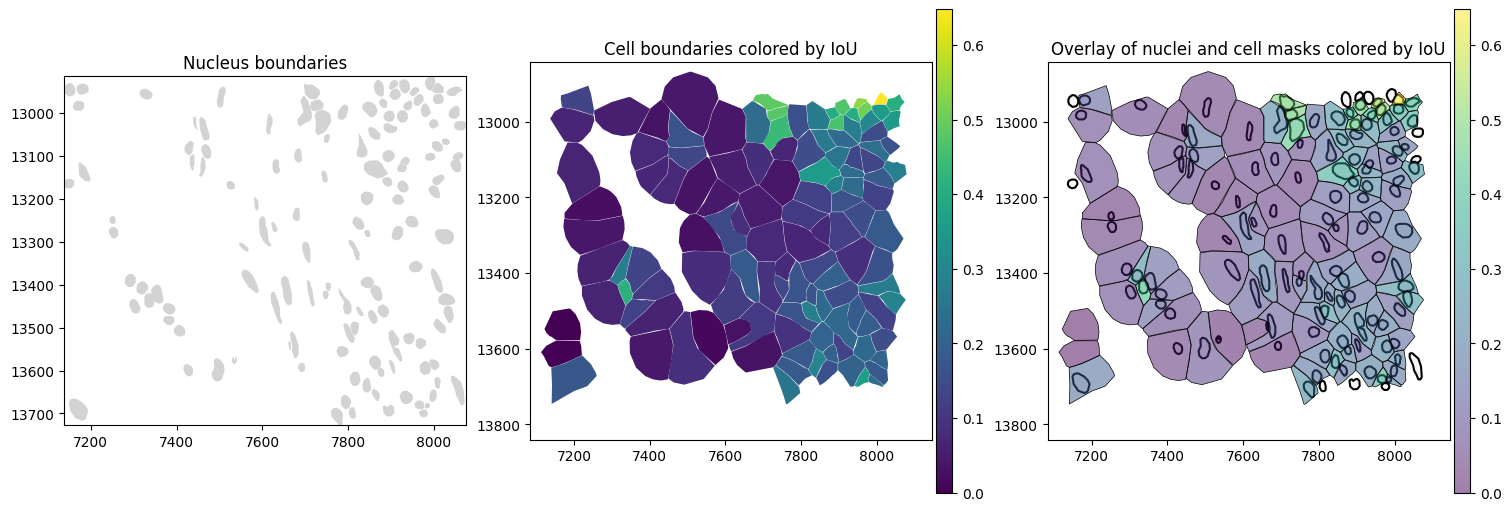
In the spatial plots, we can see that cells that have a high overlap with nuclei, also show a high IoU value.
[11]:
import matplotlib.pyplot as plt
axes = plt.subplots(1, 3, figsize=(15, 5), constrained_layout=True)[1].flatten()
# link annotations with cell boundaries
sdata.tables["table"].obs["region"] = "cell_boundaries"
sdata.set_table_annotates_spatialelement("table", region="cell_boundaries")
# plot
sdata.pl.render_shapes("nucleus_boundaries").pl.show(
ax=axes[0], title="Nucleus boundaries", coordinate_systems="global"
)
sdata.pl.render_shapes("cell_boundaries", color="IoU").pl.show(
ax=axes[1], title="Cell boundaries colored by IoU", coordinate_systems="global"
)
sdata.pl.render_shapes(
element="nucleus_boundaries",
fill_alpha=0.2,
outline_alpha=1.0,
outline_color="black",
).pl.render_shapes(
element="cell_boundaries",
color="IoU",
cmap="viridis",
fill_alpha=0.5,
outline_alpha=1.0,
outline_width=0.5,
outline_color="black",
).pl.show(ax=axes[2], title="Overlay of nuclei and cell masks colored by IoU", colorbar=True)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/spatialdata.py:511: UserWarning: Converting `region_key: region` to categorical dtype.
convert_region_column_to_categorical(table)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/_elements.py:105: UserWarning: Key `cell_boundaries` already exists. Overwriting it in-memory.
self._check_key(key, self.keys(), self._shared_keys)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/_elements.py:125: UserWarning: Key `table` already exists. Overwriting it in-memory.
self._check_key(key, self.keys(), self._shared_keys)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/_elements.py:105: UserWarning: Key `cell_boundaries` already exists. Overwriting it in-memory.
self._check_key(key, self.keys(), self._shared_keys)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/_elements.py:125: UserWarning: Key `table` already exists. Overwriting it in-memory.
self._check_key(key, self.keys(), self._shared_keys)
Expression correlation between cell and nucleus#
Expression correlation between a cell and the “most-overlapping” nucleus can be computed using compute_cell_nuc_correlation. This function will internally compute IoU using compute_cell_nuc_ious unless already present in the spatialdataobject. Currently only pearsoncorrelation is supported. More options will be added later.
[12]:
import time
import segtraq as st
[13]:
start = time.time()
corr_df = st.nc.compute_cell_nuc_correlation(sdata)
end = time.time()
print(f"Elapsed time without computing IoU: {end - start:.2f} seconds")
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/operations/aggregate.py:452: FutureWarning: The default of observed=False is deprecated and will be changed to True in a future version of pandas. Pass observed=False to retain current behavior or observed=True to adopt the future default and silence this warning.
aggregated = joined.groupby([INDEX, vk])[ONES_COLUMN].agg(agg_func).reset_index()
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/anndata/_core/aligned_df.py:68: ImplicitModificationWarning: Transforming to str index.
warnings.warn("Transforming to str index.", ImplicitModificationWarning)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/models/models.py:1144: UserWarning: Converting `region_key: region` to categorical dtype.
return convert_region_column_to_categorical(adata)
Elapsed time without computing IoU: 0.85 seconds
[14]:
corr_df
[14]:
| cell_id | best_nuc_id | IoU | correlation | |
|---|---|---|---|---|
| 0 | 4613 | 4613 | 0.094093 | 0.272743 |
| 1 | 4614 | 4614 | 0.207238 | 0.878210 |
| 2 | 4615 | 4615 | 0.178686 | 0.739670 |
| 3 | 4616 | 4616 | 0.292909 | 0.779925 |
| 4 | 4617 | 4617 | 0.161824 | 0.910924 |
| ... | ... | ... | ... | ... |
| 117 | 80759 | 80759 | 0.160499 | 0.812877 |
| 118 | 80760 | 80760 | 0.128202 | 0.914975 |
| 119 | 80761 | 80761 | 0.288906 | 0.913159 |
| 120 | 80762 | 80762 | 0.180239 | 0.913756 |
| 121 | 80763 | 80763 | 0.269007 | 0.733573 |
122 rows × 4 columns
We will store the results in the .obs of the Table within the sdata object for plotting.
[15]:
corr_df = corr_df.set_index("cell_id")
sdata["table"].obs["pearson_corr"] = sdata["table"].obs["cell_id"].map(corr_df["correlation"])
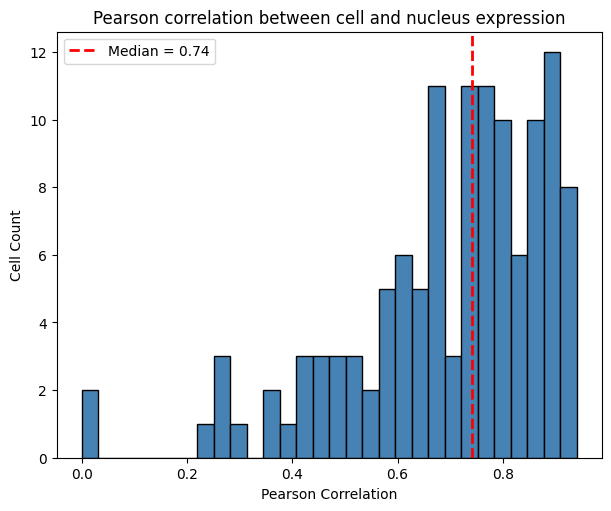
The histogram below shows the distribution of Pearson correlation values across cells. It is right-skewed, with a median of 0.74. This provides intuition about possible spatial spillover: cells may be contaminated by neighboring cells, assuming that nuclei capture expression with less contamination due to their smaller radius.
[16]:
# Prepare corr values
obs = sdata["table"].obs
corr = obs["pearson_corr"]
median_corr = np.median(corr)
# Set up figure with histogram
fig, ax = plt.subplots(figsize=(6, 5), constrained_layout=True)
# Plot histogram
ax.hist(corr, bins=30, color="steelblue", edgecolor="black")
# Add median line
ax.axvline(
median_corr,
color="red",
linestyle="--",
linewidth=2,
label=f"Median = {median_corr:.2f}",
)
# Decorate
ax.set_title("Pearson correlation between cell and nucleus expression")
ax.set_xlabel("Pearson Correlation")
ax.set_ylabel("Cell Count")
ax.legend()
plt.show()
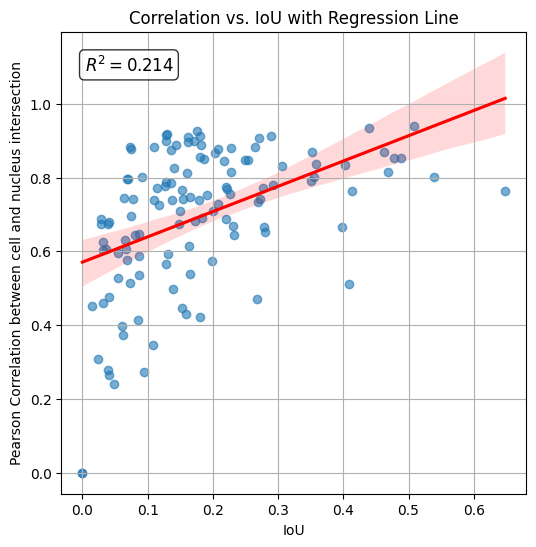
The calculated R2 (coefficient of determination) is 0.214 indicating that correlation between cell and nucleus increases with their IoU.
[17]:
import seaborn as sns
from scipy.stats import linregress
# Extract the relevant columns, excluding NaNs
df = obs[["IoU", "pearson_corr"]].dropna()
# Compute regression metrics
slope, intercept, r_value, p_value, std_err = linregress(df["IoU"], df["pearson_corr"])
r_squared = r_value**2
# Create the plot
plt.figure(figsize=(6, 6))
ax = sns.regplot(
data=df,
x="IoU",
y="pearson_corr",
scatter_kws={"alpha": 0.6},
line_kws={"color": "red"},
ci=95,
)
# Overlay the R² value
ax.text(
0.05,
0.95,
f"$R^2 = {r_squared:.3f}$",
transform=ax.transAxes,
verticalalignment="top",
fontsize=12,
bbox=dict(boxstyle="round,pad=0.3", facecolor="white", alpha=0.8),
)
ax.set_xlabel("IoU")
ax.set_ylabel("Pearson Correlation between cell and nucleus intersection")
ax.set_title("Correlation vs. IoU with Regression Line")
ax.grid(True)
plt.show()
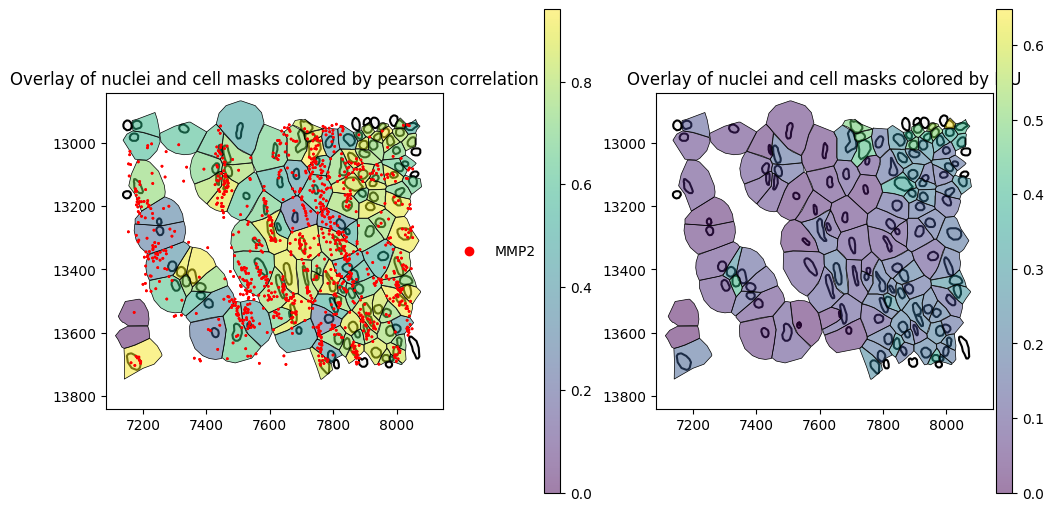
The spatial plot below overlays nucleus boundaries, cell masks colored by Pearson correlation, and transcripts. Cells whose transcripts are distributed across the nucleus region tend to have higher correlation with the nucleus, while cells whose transcripts are localized outside the nuclear region—potentially due to spillover from neighboring cells—tend to exhibit lower correlation.
[18]:
# link annotations with cell boundaries
sdata.tables["table"].obs["region"] = "cell_boundaries"
sdata.set_table_annotates_spatialelement("table", region="cell_boundaries")
axes = plt.subplots(1, 2, figsize=(10, 5), constrained_layout=True)[1].flatten()
sdata.pl.render_shapes(
element="nucleus_boundaries",
fill_alpha=0.2,
outline_alpha=1.0,
outline_color="black",
).pl.render_shapes(
element="cell_boundaries",
color="pearson_corr",
cmap="viridis",
fill_alpha=0.5,
outline_alpha=1.0,
outline_width=0.5,
outline_color="black",
).pl.render_points(
"transcripts",
color="feature_name",
groups="MMP2",
palette="red",
).pl.show(
ax=axes[0],
title="Overlay of nuclei and cell masks colored by pearson correlation",
colorbar=True,
figsize=(6, 6),
)
sdata.pl.render_shapes(
element="nucleus_boundaries",
fill_alpha=0.2,
outline_alpha=1.0,
outline_color="black",
).pl.render_shapes(
element="cell_boundaries",
color="IoU",
cmap="viridis",
fill_alpha=0.5,
outline_alpha=1.0,
outline_width=0.5,
outline_color="black",
).pl.show(
ax=axes[1],
title="Overlay of nuclei and cell masks colored by IoU",
colorbar=True,
figsize=(6, 6),
)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/spatialdata.py:511: UserWarning: Converting `region_key: region` to categorical dtype.
convert_region_column_to_categorical(table)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/_elements.py:105: UserWarning: Key `cell_boundaries` already exists. Overwriting it in-memory.
self._check_key(key, self.keys(), self._shared_keys)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/_elements.py:125: UserWarning: Key `table` already exists. Overwriting it in-memory.
self._check_key(key, self.keys(), self._shared_keys)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/anndata/_core/anndata.py:381: FutureWarning: The dtype argument is deprecated and will be removed in late 2024.
warnings.warn(
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/anndata/_core/aligned_df.py:68: ImplicitModificationWarning: Transforming to str index.
warnings.warn("Transforming to str index.", ImplicitModificationWarning)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/_elements.py:115: UserWarning: Key `transcripts` already exists. Overwriting it in-memory.
self._check_key(key, self.keys(), self._shared_keys)
INFO input has more than 103 categories. Uniform 'grey' color will be used for all categories.
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata_plot/pl/utils.py:775: FutureWarning: The default value of 'ignore' for the `na_action` parameter in pandas.Categorical.map is deprecated and will be changed to 'None' in a future version. Please set na_action to the desired value to avoid seeing this warning
color_vector = color_source_vector.map(color_mapping)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata_plot/pl/render.py:682: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap', 'norm' will be ignored
_cax = ax.scatter(
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/_elements.py:105: UserWarning: Key `cell_boundaries` already exists. Overwriting it in-memory.
self._check_key(key, self.keys(), self._shared_keys)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/_elements.py:125: UserWarning: Key `table` already exists. Overwriting it in-memory.
self._check_key(key, self.keys(), self._shared_keys)
Correlation between Intersection and Remainder Parts of the Cell#
The function compute_correlation_between_parts() computes the Pearson correlation between the spatial transcript feature counts within the intersection of a cell and its matched nucleus, and the remainder of the cell. It returns NaN when the regions have no overlapping transcripts or lack sufficient variability. A low correlation may indicate that the cell boundary extension captures neighborhood or ambient signal rather than true intracellular expression—a concern similarly highlighted
by segmentation benchmarks such as Baysor. This is based on the assumption that transcripts are homogeneously distributed within the cell and not localized in specific subcellular regions.
[19]:
start = time.time()
corr_parts_df = st.nc.compute_correlation_between_parts(sdata)
end = time.time()
print(f"Elapsed time without computing IoU: {end - start:.2f} seconds")
Computing correlations: 122it [00:02, 59.79it/s]
Elapsed time without computing IoU: 2.07 seconds
[20]:
len(corr_parts_df) == len(sdata.tables["table"].obs)
[20]:
True
We will store the results in the .obs of the Table within the sdata object for plotting.
[32]:
corr_parts_df = corr_parts_df.set_index("cell_id")
sdata["table"].obs["pearson_corr_parts"] = sdata["table"].obs["cell_id"].map(corr_parts_df["correlation_parts"])
[33]:
sdata["table"].obs
[33]:
| cell_id | transcript_counts | control_probe_counts | control_codeword_counts | total_counts | cell_area | nucleus_area | region | IoU | best_nuc_id | pearson_corr | pearson_corr_parts | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4612 | 4613 | 210 | 0 | 0 | 210 | 272.066406 | 27.319531 | cell_boundaries | 0.094093 | 4613 | 0.272743 | 0.113081 |
| 4613 | 4614 | 192 | 0 | 0 | 192 | 154.208594 | 33.280156 | cell_boundaries | 0.207238 | 4614 | 0.878210 | 0.529910 |
| 4614 | 4615 | 174 | 0 | 0 | 174 | 170.193906 | 31.744844 | cell_boundaries | 0.178686 | 4615 | 0.739670 | 0.289943 |
| 4615 | 4616 | 70 | 0 | 0 | 70 | 85.796875 | 26.235781 | cell_boundaries | 0.292909 | 4616 | 0.779925 | 0.258221 |
| 4616 | 4617 | 85 | 0 | 0 | 85 | 101.737031 | 16.843281 | cell_boundaries | 0.161824 | 4617 | 0.910924 | 0.761505 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 80758 | 80759 | 227 | 0 | 0 | 227 | 256.984219 | 42.221094 | cell_boundaries | 0.160499 | 80759 | 0.812877 | 0.628438 |
| 80759 | 80760 | 198 | 0 | 0 | 198 | 222.800938 | 30.028906 | cell_boundaries | 0.128202 | 80760 | 0.914975 | 0.896952 |
| 80760 | 80761 | 110 | 0 | 0 | 110 | 111.355313 | 33.144688 | cell_boundaries | 0.288906 | 80761 | 0.913159 | 0.751234 |
| 80761 | 80762 | 150 | 0 | 0 | 150 | 322.189844 | 60.644844 | cell_boundaries | 0.180239 | 80762 | 0.913756 | 0.786104 |
| 80762 | 80763 | 109 | 0 | 0 | 109 | 152.763594 | 41.769531 | cell_boundaries | 0.269007 | 80763 | 0.733573 | 0.360049 |
122 rows × 12 columns
[23]:
sdata["table"].obs["pearson_corr_parts"].isna().sum()
[23]:
np.int64(2)
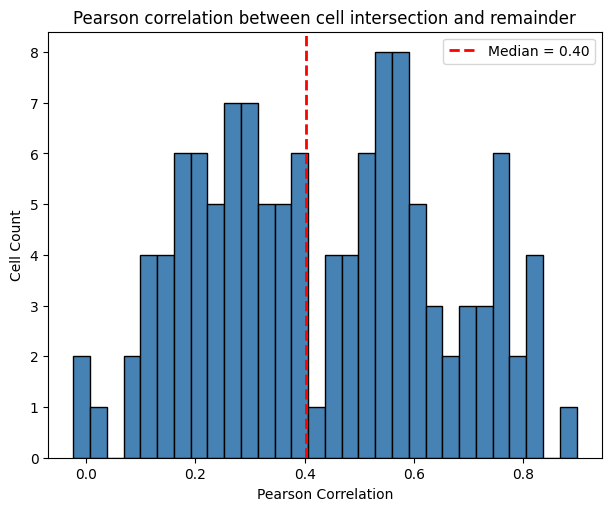
The histogram below shows that the Pearson correlation between the intersection and remainder region of the cell is 0.4.
[24]:
obs = sdata["table"].obs
# Prepare corr_parts values
corr = obs["pearson_corr_parts"]
median_corr_parts = corr.median()
# Set up figure with histogram
fig, ax = plt.subplots(figsize=(6, 5), constrained_layout=True)
# Plot histogram
ax.hist(corr, bins=30, color="steelblue", edgecolor="black")
# Add median line
ax.axvline(
median_corr_parts,
color="red",
linestyle="--",
linewidth=2,
label=f"Median = {median_corr_parts:.2f}",
)
# Decorate
ax.set_title("Pearson correlation between cell intersection and remainder")
ax.set_xlabel("Pearson Correlation")
ax.set_ylabel("Cell Count")
ax.legend()
plt.show()
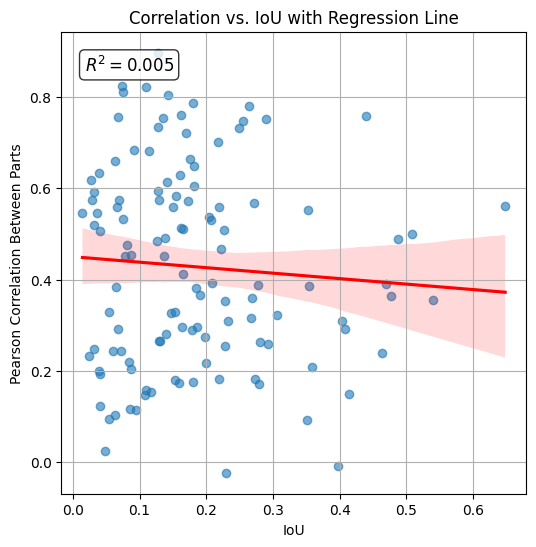
The histogram below visualizes the Pearson correlation between transcript counts in the intersection and remainder of each cell, plotted against the Intersection over Union (IoU) with the nucleus. The calculated R2 (coefficient of determination) value is 0.005, indicating that IoU explains less than 1% of the variance in correlation. This negligible relationship suggests that normalization of the correlation metric by IoU is not necessary.
[25]:
import seaborn as sns
# Extract the relevant columns, excluding NaNs
df = obs[["IoU", "pearson_corr_parts"]].dropna()
# Compute regression metrics
slope, intercept, r_value, p_value, std_err = linregress(df["IoU"], df["pearson_corr_parts"])
r_squared = r_value**2
# Create the plot
plt.figure(figsize=(6, 6))
ax = sns.regplot(
data=df,
x="IoU",
y="pearson_corr_parts",
scatter_kws={"alpha": 0.6},
line_kws={"color": "red"},
ci=95,
)
# Overlay the R² value
ax.text(
0.05,
0.95,
f"$R^2 = {r_squared:.3f}$",
transform=ax.transAxes,
verticalalignment="top",
fontsize=12,
bbox=dict(boxstyle="round,pad=0.3", facecolor="white", alpha=0.8),
)
ax.set_xlabel("IoU")
ax.set_ylabel("Pearson Correlation Between Parts")
ax.set_title("Correlation vs. IoU with Regression Line")
ax.grid(True)
plt.show()
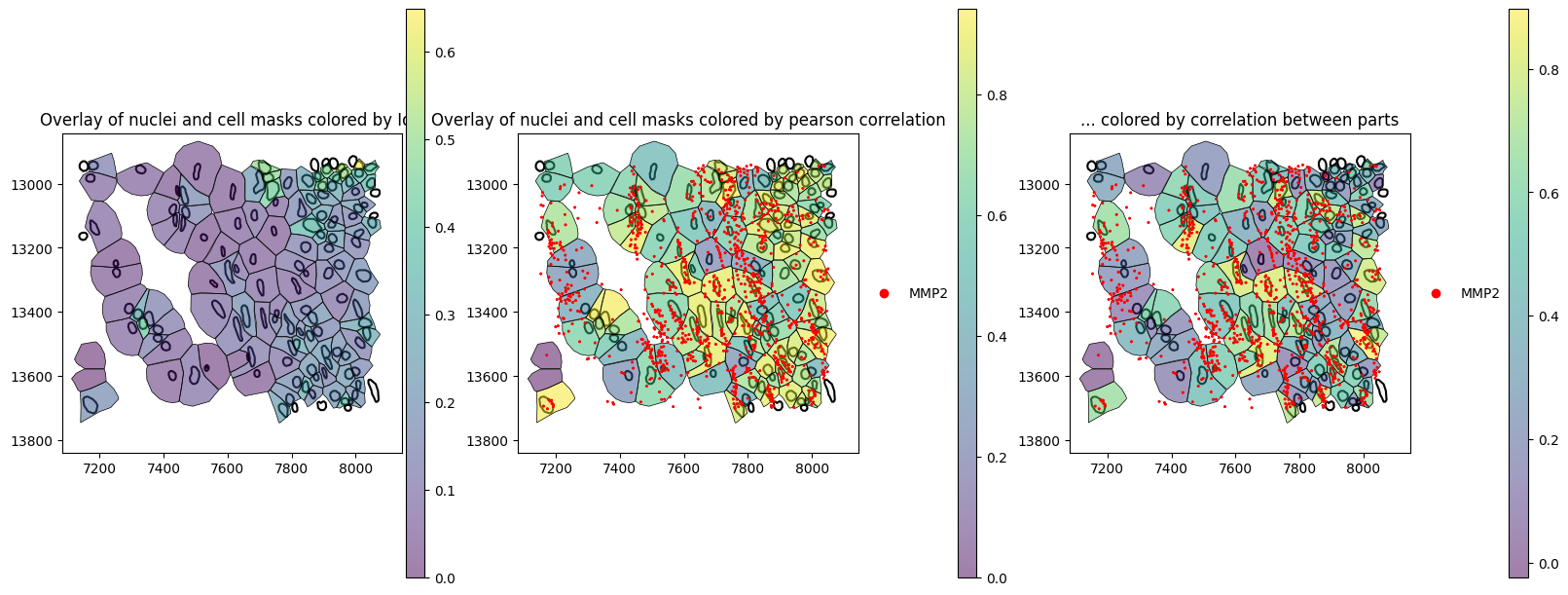
Because spatialdata-plot plots all boundaries in grey, if there are NaN values in the annotation (describe before), we will copy the pearson_corr_parts column and set NaNvalues to 0.0just for plotting. We will try to solve this issue later.
[26]:
sdata["table"].obs["pearson_corr_parts_plot"] = (
sdata["table"].obs["cell_id"].map(corr_parts_df["correlation_parts"])
) # TODO
sdata["table"].obs.loc[sdata["table"].obs["pearson_corr_parts_plot"].isna(), "pearson_corr_parts_plot"] = 0.0
The spatial plots below shows the spatial distribution of computed correlations. The Pearson correlation between parts can be a measure of how much neighboring signal is captured.
[27]:
# link annotations with cell boundaries
sdata.tables["table"].obs["region"] = "cell_boundaries"
sdata.set_table_annotates_spatialelement("table", region="cell_boundaries")
axes = plt.subplots(1, 3, figsize=(16, 6), constrained_layout=True)[1].flatten()
sdata.pl.render_shapes(
element="nucleus_boundaries",
fill_alpha=0.2,
outline_alpha=1.0,
outline_color="black",
).pl.render_shapes(
element="cell_boundaries",
color="IoU",
cmap="viridis",
fill_alpha=0.5,
outline_alpha=1.0,
outline_width=0.5,
outline_color="black",
).pl.show(
ax=axes[0],
title="Overlay of nuclei and cell masks colored by IoU",
colorbar=True,
figsize=(6, 6),
)
sdata.pl.render_shapes(
element="nucleus_boundaries",
fill_alpha=0.2,
outline_alpha=1.0,
outline_color="black",
).pl.render_shapes(
element="cell_boundaries",
color="pearson_corr",
cmap="viridis",
fill_alpha=0.5,
outline_alpha=1.0,
outline_width=0.5,
outline_color="black",
).pl.render_points(
"transcripts",
color="feature_name",
groups="MMP2",
palette="red",
).pl.show(
ax=axes[1],
title="Overlay of nuclei and cell masks colored by pearson correlation",
colorbar=True,
figsize=(6, 6),
)
sdata.pl.render_shapes(
element="nucleus_boundaries",
fill_alpha=0.2,
outline_alpha=1.0,
outline_color="black",
).pl.render_shapes(
element="cell_boundaries",
color="pearson_corr_parts_plot",
cmap="viridis",
fill_alpha=0.5,
outline_alpha=1.0,
outline_width=0.5,
outline_color="black",
).pl.render_points(
"transcripts",
color="feature_name",
groups="MMP2",
palette="red",
).pl.show(
ax=axes[2],
title="... colored by correlation between parts",
colorbar=True,
figsize=(6, 6),
)
# remove column again
sdata["table"].obs.drop(columns=["pearson_corr_parts_plot"], inplace=True)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/spatialdata.py:511: UserWarning: Converting `region_key: region` to categorical dtype.
convert_region_column_to_categorical(table)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/_elements.py:105: UserWarning: Key `cell_boundaries` already exists. Overwriting it in-memory.
self._check_key(key, self.keys(), self._shared_keys)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/_elements.py:125: UserWarning: Key `table` already exists. Overwriting it in-memory.
self._check_key(key, self.keys(), self._shared_keys)
INFO input has more than 103 categories. Uniform 'grey' color will be used for all categories.
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/_elements.py:105: UserWarning: Key `cell_boundaries` already exists. Overwriting it in-memory.
self._check_key(key, self.keys(), self._shared_keys)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/_elements.py:125: UserWarning: Key `table` already exists. Overwriting it in-memory.
self._check_key(key, self.keys(), self._shared_keys)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/anndata/_core/anndata.py:381: FutureWarning: The dtype argument is deprecated and will be removed in late 2024.
warnings.warn(
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/anndata/_core/aligned_df.py:68: ImplicitModificationWarning: Transforming to str index.
warnings.warn("Transforming to str index.", ImplicitModificationWarning)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/_elements.py:115: UserWarning: Key `transcripts` already exists. Overwriting it in-memory.
self._check_key(key, self.keys(), self._shared_keys)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata_plot/pl/utils.py:775: FutureWarning: The default value of 'ignore' for the `na_action` parameter in pandas.Categorical.map is deprecated and will be changed to 'None' in a future version. Please set na_action to the desired value to avoid seeing this warning
color_vector = color_source_vector.map(color_mapping)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata_plot/pl/render.py:682: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap', 'norm' will be ignored
_cax = ax.scatter(
INFO input has more than 103 categories. Uniform 'grey' color will be used for all categories.
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/_elements.py:105: UserWarning: Key `cell_boundaries` already exists. Overwriting it in-memory.
self._check_key(key, self.keys(), self._shared_keys)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/_elements.py:125: UserWarning: Key `table` already exists. Overwriting it in-memory.
self._check_key(key, self.keys(), self._shared_keys)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/anndata/_core/anndata.py:381: FutureWarning: The dtype argument is deprecated and will be removed in late 2024.
warnings.warn(
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/anndata/_core/aligned_df.py:68: ImplicitModificationWarning: Transforming to str index.
warnings.warn("Transforming to str index.", ImplicitModificationWarning)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata/_core/_elements.py:115: UserWarning: Key `transcripts` already exists. Overwriting it in-memory.
self._check_key(key, self.keys(), self._shared_keys)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata_plot/pl/utils.py:775: FutureWarning: The default value of 'ignore' for the `na_action` parameter in pandas.Categorical.map is deprecated and will be changed to 'None' in a future version. Please set na_action to the desired value to avoid seeing this warning
color_vector = color_source_vector.map(color_mapping)
/home/lazic/miniforge3/envs/sc_analysis_sdata/lib/python3.10/site-packages/spatialdata_plot/pl/render.py:682: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap', 'norm' will be ignored
_cax = ax.scatter(
[ ]: